Topics
2022.10.31 circular No.19
新しい時間分解分光計の製作学習院大学理学部化学科
岩田 耕一
筆者の専門は分光実験である。新しい測定ができる分光計をつくって、その分光計で新しい現象を観測するのが研究の目標になっている。今回は、現在研究室で稼働しているいくつかのオリジナルな分光計について紹介してみたい。
2.ピコ秒時間分解ラマン分光計
ラマン分光法では、物質からの光の非弾性散乱を測定する。光が散乱されるときは、大部分の光が入射光と同じ波長のまま散乱されるが、違った波長で散乱される光がまれにある。このような非弾性散乱光のことをラマン散乱光と呼ぶ。通常のラマン散乱(「自発ラマン散乱」と呼ぶ)は自然放出に対応していて、ラマン散乱光はあらゆる方向に放射される。このラマン散乱光の一部をレンズで集めて分光器に導き、波長分散させてから検出するのがラマン分光法である。
ラマン散乱が起こるのは「一瞬」なので、励起光がパルス光の場合はラマン散乱光もそのパルス光の持続時間の間にしか発生しない。ピコ秒時間分解ラマン分光法では、励起光(「プローブ光」)にピコ秒のパルス光を使うことで、測定対象の構造や状態に関するピコ秒の間の情報を得ている。ピコ秒時間分解ラマン分光法は、1990年代にレーザー光源や検出器の進歩とともに急速に発展して、測定法がほぼ確立した(と思われていた)。ただし、ラマン分光計の光源に適した繰り返し周波数(kHz程度)や平均出力(mW程度)のピコ秒パルスを出力できるレーザーは実際には安定性に難があることが多く、ラマン分光計の主要構成要素であるピコ秒レーザーを毎日安定に稼働させることは現在でも容易でない。これに対して、チタンサファイア再生増幅器を基本とするパルス幅100 fs、繰り返し周波数1 kHz程度のフェムト秒レーザーの出力安定性は、ピコ秒レーザーに比べてはるかに優れている。筆者らは、このフェムト秒レーザーを光源とすることで、従来よりも格段に安定なピコ秒時間分解ラマン分光計を作れるのではないかと考えた。
フェムト秒の光パルスを延伸したピコ秒光パルスは、ラマンスペクトルを測定するときにプローブ光として用いられる。一方で、時間分解ラマンスペクトルを測定するときは、試料に照射して電子励起や化学反応などを引き起こす「ポンプ光」も必要である。ピコ秒の時間分解測定のときには、ポンプ光とプローブ光のそれぞれが試料に到着するときの時間差のゆらぎ(「タイミングジッター」)も小さくなくてはならない。
フェムト秒の光パルスをピコ秒に延伸することは、実はそれほど容易でない。筆者らは、フェムト秒光パルスをピコ秒光パルスに変換するために、「体積グレーティングノッチフィルター」を利用することにした。体積グレーティングノッチフィルターは、ガラス素材の中に回折格子が焼き付けられた1枚の光学フィルターである。このフィルターは特定の波長の光のみを狭帯域で除去して残りの光は透過する画期的な光学素子で、ラマン分光測定においてレイリー散乱光を除去するための高性能のノッチフィルターとして2010年ころから急速に利用が進んだ。ラマンスペクトルの測定時には体積グレーティングノッチフィルターを透過した光が分光器に入っていくのだが、筆者らは反射した狭帯域の光の方を利用することにした。光の波としての性質から、体積グレーティングノッチフィルターで反射された波数幅が数 cm-1しかない光は、時間の軸でみるとピコ秒のパルスになる。フィルターを1枚使うのみでフェムト秒の光パルスをピコ秒の光パルスに変換できることになる。フェムト秒のチタンサファイアレーザーを光源としたピコ秒時間分解ラマン分光計[1]では、光源からの出力であるフェムト秒パルスを体積グレーティングノッチフィルターで反射することで、ピコ秒のプローブ光を作っている(図1)。ポンプ光にはプローブ光のような小さいエネルギー幅(波数幅)は必要ないから、波長変換したフェムト秒の光パルスを延伸することなくそのまま使っている。ポンプ光とプローブ光はもともと同じレーザー光源からの出力光であるから、両者の間のタイミングジッターは無視できる程度に小さい。得られたプローブ光のスペクトル幅は6.0 cm-1であり、電子励起されたtrans-スチルベン分子の過渡吸収の立ち上がりから見積もった時間幅は3.2 psだった。
光パルスの波数幅と時間幅の両方を無限に小さくすることはできず、両者の積はある一定値よりも必ず大きくなる。積が最小値になるとき、その波は「フーリエ変換限界」にあるが、新しい方法で作った図1のピコ秒時間分解ラマン分光計でのプローブ光の波数幅と時間幅の積は(光の電場ではなくエネルギーで評価した)フーリエ変換限界の1.3倍であった。体積グレーティングノッチフィルターを使うことで、フーリエ変換限界にきわめて近いピコ秒のプローブ光パルスを作ることができたことになる。
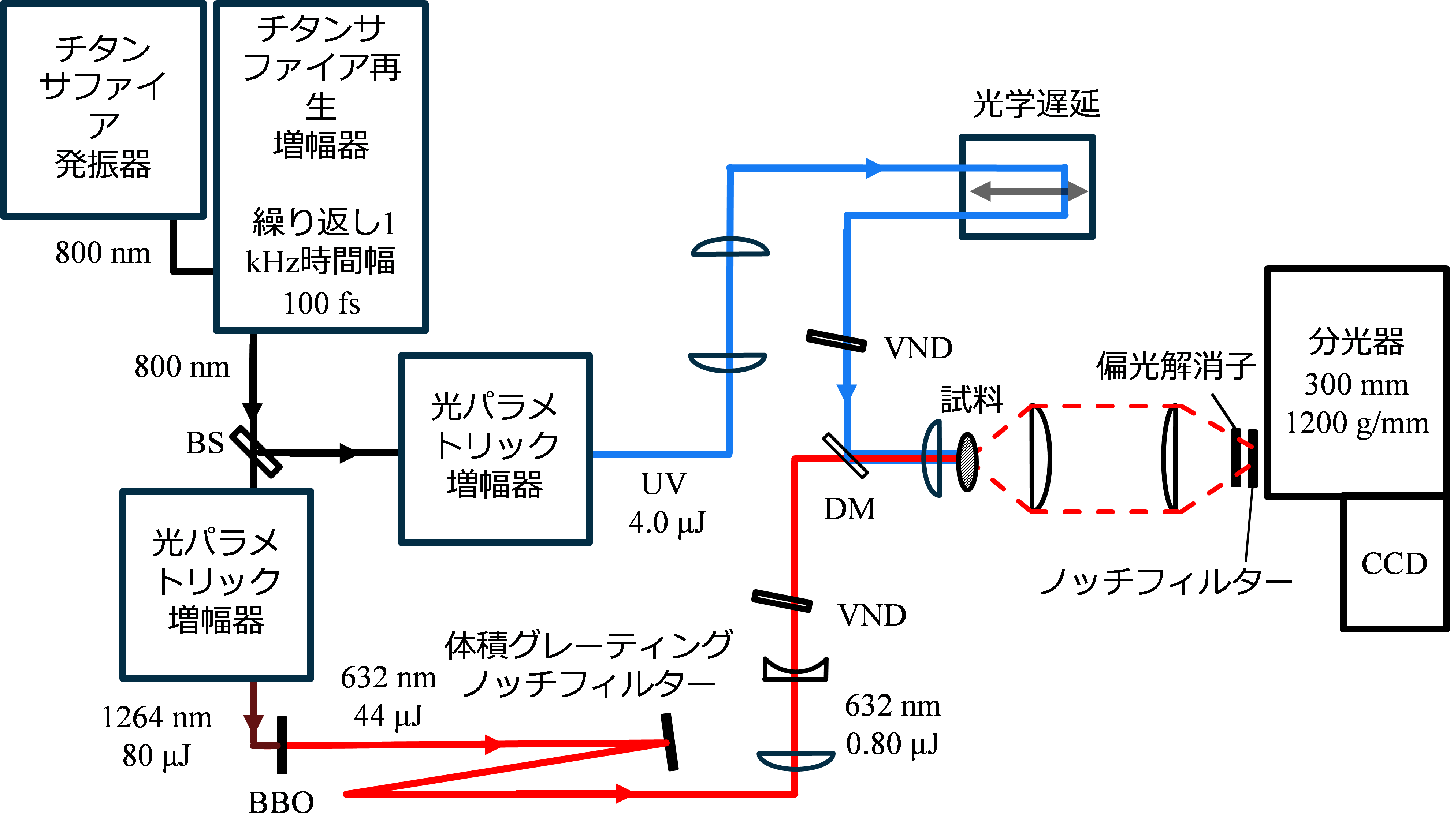
図1 ピコ秒時間分解ラマン分光計のブロック図

図2 ピコ秒プローブ光強度の時間変化
新しい方式の装置を作った動機は出力安定性に優れた時間分解ラマン分光計を使いたいというものだった。プローブ光の出力安定性を実際に測定してみると(図2)、5000秒間でのプローブ光出力の揺らぎは二乗平均平方根(RMS)で0.8%となった[1]。この値は筆者らが従来使っていたピコ秒時間分解ラマン分光計よりも10倍以上優れている。新たなピコ秒時間分解ラマン分光計で測定した最低励起1重項(S1)状態のtrans-スチルベンの時間分解ラマンスペクトル(図3)では、それぞれの遅延時間でのラマンスペクトルを測定するときに1分間しか必要としなかった。この装置を使うことで質の高い時間分解ラマンスペクトルを迅速に測れることを示すことができた。
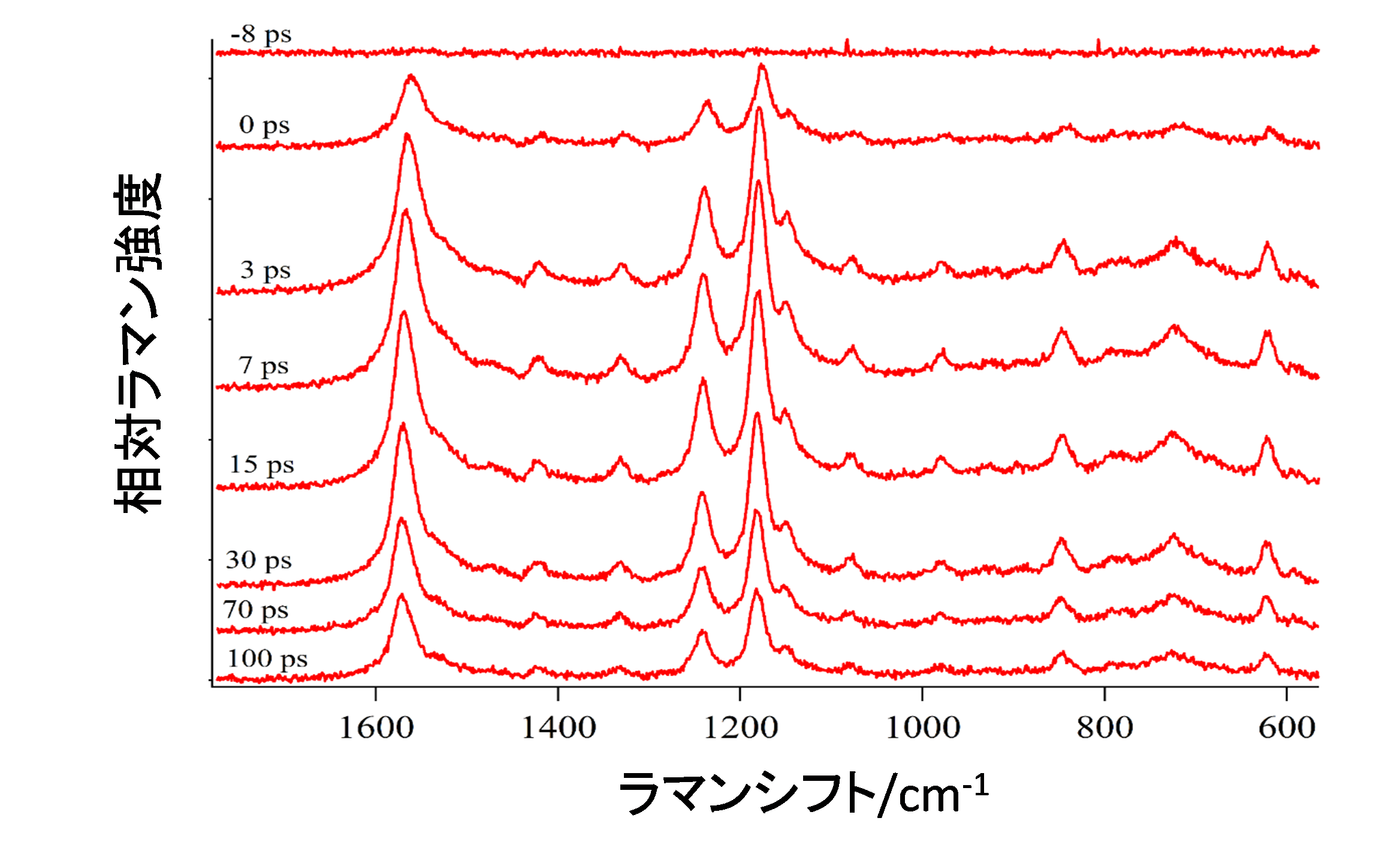
図3 ヘプタン溶液中で測定したS1 trans-スチルベンの時間分解ラマンスペクトル
筆者らは、かつてC2mimTf2N、C4mimTf2N、C4mimPF6、C5mimTf2N、C6mimTf2N、C8mimTf2NおよびbmpyTf2N中に溶解したtrans-スチルベン分子を余剰エネルギーとともにS1状態に光励起して、その後の冷却過程をピコ秒時間分解ラマン分光法で追跡した。その結果を熱拡散方程式によるモデル計算と比較することで、イオン液体中に形成される局所構造の大きさが10から100 nmであると推定することができた[2]。従来よりも格段に安定なラマン分光計を使って精密な測定を行えば、イオン液体の構造やイオン液体中で起こる化学反応についてより精密な描像を得ることができるだろう。 3.フェムト秒時間分解近赤外吸収分光計とフェムト秒時間分解近赤外非線形ラマン分光計
化学反応は価電子の組み換えといえる。電子は化学反応にともなって始原系の安定な場所を離れて生成系の安定な場所へと移る。この過程で、電子は原子核からの引力から部分的に自由になり得る。このような「束縛の弱い電子」は、紫外・可視域よりも遷移エネルギーが小さい近赤外領域に吸収を示すことが期待される。溶媒和電子やC=C共役系の励起状態、伝導電帯中の電子などがこの例といえる。束縛の弱い電子を時間分解近赤外吸収分光法で観測できれば、化学反応の進行について新たな視点から理解できるようになるだろう。
時間分解吸収分光法は、1940年代に可視領域[3]と中赤外領域[4、5]で始まった。それ以来、特に可視領域の時間分解分光法がさまざまな研究に使われて、化学反応の中間体など短寿命の分子種に関する多くの知見をもたらしてきた。CCD検出器に代表されるマルチチャンネル検出器が登場してからは、時間分解吸収測定の効率が飛躍的に高まった。しかし、これらのマルチチャンネル検出器の多くはシリコン製であり、そのバンドギャップに相当する1000 nmよりも長波長の光を検出することができない。筆者らは、InGaAsのアレイ検出器を利用することで、波長1000から1500 nmで測定可能なフェムト秒時間分解近赤外吸収分光計を製作した。この分光計を用いて、ビアントリル[7]やβ-カロテン[8]、イオン液体中や通常の溶媒中での溶媒和電子[9、10]の挙動を解明してきた。
ラマンスペクトルの測定において励起光の波長が測定対象の分子の吸収帯の波長に近づくと、ラマン散乱の確率が103かそれ以上に増大する。「共鳴ラマン効果」と呼ばれるこの現象を利用すると、測定試料中に多くの種類の分子が混在していても狙った分子あるいは分子の電子励起状態のラマンスペクトルを選択的に測定することができる[11]。近赤外領域の励起光を使って共鳴ラマンスペクトルを測定することができれば、近赤外領域に吸収をもつ分子種を選択的に検出できるだろう。しかし、前節で記したように、1000から1500 nmの波長領域にはラマン分光測定に適した検出器がない。微弱光検出に適した光検出器がないことが近赤外領域でのラマン分光測定を制約していた。筆者らが近赤外領域での時間分解吸収測定で利用しているInGaAs検出器は感度の高い検出器ではあるが、冷却されたCCD検出器に比べると雑音が数桁大きい。自発ラマン分光法は一種の発光分光法であり、試料が発光しないかぎり背景光はない。スペクトルのSN比を決める雑音としては、検出器の雑音が支配的である。自発ラマン散乱を測定するためには、低雑音の検出器を利用できることが必須なのである。
自発ラマン散乱は光の自然放出に対応する光学過程であるが、それに対して光の誘導放出に対応するラマン過程もある。後者は、「誘導ラマン散乱」と呼ばれる。誘導ラマン散乱は「非線形ラマン散乱」の一種であり、その確率は入射光の電場強度のみでなく誘導放出を引き起こす光の電場強度にも依存する。誘導ラマン散乱を測定するときは、試料にラマン励起とプローブの2種類の光を同時に入射する。ラマン励起は自発ラマン散乱測定のときの励起光と同じ役割を果たす。プローブ光は、誘導放出によってラマン散乱による発光を引き起こす。誘導ラマンスペクトルを測定すると、試料を通過した後のプローブ光の強度がラマン励起光からラマンシフトの分だけ離れた波長で増加する。誘導ラマン散乱の測定のときはラマン励起光の有無によるプローブ光の変化を測定するから、検出器はいつもプローブ光を検出している。プローブ光の強度によっては、スペクトルの雑音は検出器雑音よりもプローブ光のショットノイズが支配的になる。低雑音の検出器の利用を期待できない近赤外領域では、誘導ラマン効果を使うことでSN比が大きいラマンスペクトルを測定できる可能性がある。筆者らは、前述のフェムト秒時間分解近赤外吸収分光計に近赤外領域のラマン励起光を付け加えてフェムト秒時間分解近赤外非線形ラマン分光計を製作した(図4)[12]。この分光計では、過渡吸収測定で用いる「ポンプ光」と「プローブ光」の2種類のフェムト光パルスに加えて、ピコ秒の「ラマン励起光」の合計3種類の光を試料に照射する。最初に述べたピコ秒時間分解ラマン分光計でのプローブ光に相当するラマン励起光の波長は1190 nmである。この光を作るためには、ピコ秒時間分解ラマン分光計と同様に体積グレーティングノッチフィルターを利用した。
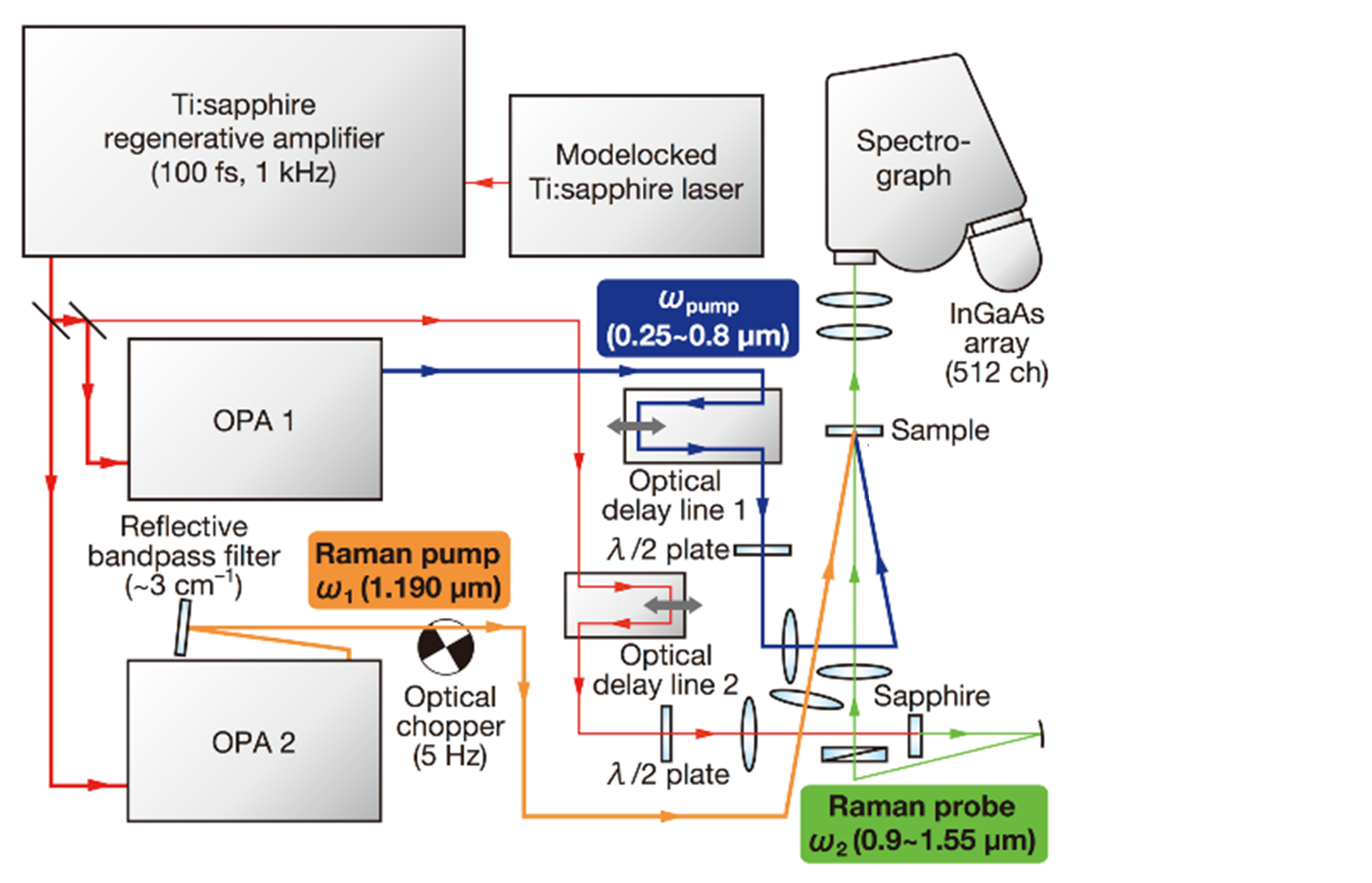
図4 フェムト秒時間分解近赤外非線形ラマン分光計のブロック図
フェムト秒時間分解近赤外非線形ラマン分光計でラマン励起光を遮断すれば、通常の時間分解近赤外吸収スペクトルを測定できる。このようにして測定したβ-カロテンの時間分解近赤外吸収スペクトルとラマン励起光も使って測定した時間分解近赤外誘導ラマンスペクトルを図5に示す[8]。励起後にすぐβ-カロテンS2状態を始状態とする過渡吸収帯が1000 nm付近に観測され、0.61 psからはS1状態を始状態とする1200 nm付近および1500 nm付近の吸収帯が現れる(図5A)。β-カロテンのS2状態は基底状態からの遷移が光学許容であり、β-カロテンの赤い色の起源である500 nm付近の強い吸収は基底状態からS2状態への遷移によるものである。一方で、基底状態からS1状態への遷移は光学禁制である。S1状態はカロテノイドの高速な無輻射失活の原因となる電子状態であるが、吸収スペクトルではS1状態を観測できないのでそのエネルギーを決めることは容易でなかった。近赤外領域での時間分解吸収スペクトルを測定することで、S1状態の正確なエネルギーを特定することができたのである。さらに、S2状態からS1状態への内部転換は190 fsで進行することがわかった。これらの電子吸収に共鳴した時間分解ラマンスペクトルでは、S2状態とS1状態のC=C伸縮振動がそれぞれ1556と1791 cm-1に観測されている(図5B)。このうち、1791 cm-1のラマンバンドは時間の経過とともにその位置と形状が大きく変化する。この変化から、S1状態での振動緩和は0.9から1.2 psで進行することが分かった。ここで述べるように、光を吸収したβ-カロテンに起こる現象を詳細に調べることが可能になった。β-カロテン末端のヨノン環にOH基あるいはC=O基を導入した誘導体であるゼアキサンチン、カンタキサンチンおよびアセタキサンチンでの測定結果もあわせて4種類のカロテノイドを比較検討すると、S2状態とS1状態のエネルギー差と前者から後者への内部転換の速度、S1状態でのC=C伸縮振動の波数および振動緩和速度のすべてにおいて、ヨノン環におけるC=O基の有無によって違いがあることが分かった。これらの違いはOH基の有無には影響されなかった[13]。これらの予期しなかった現象の起源はまだ分からない。
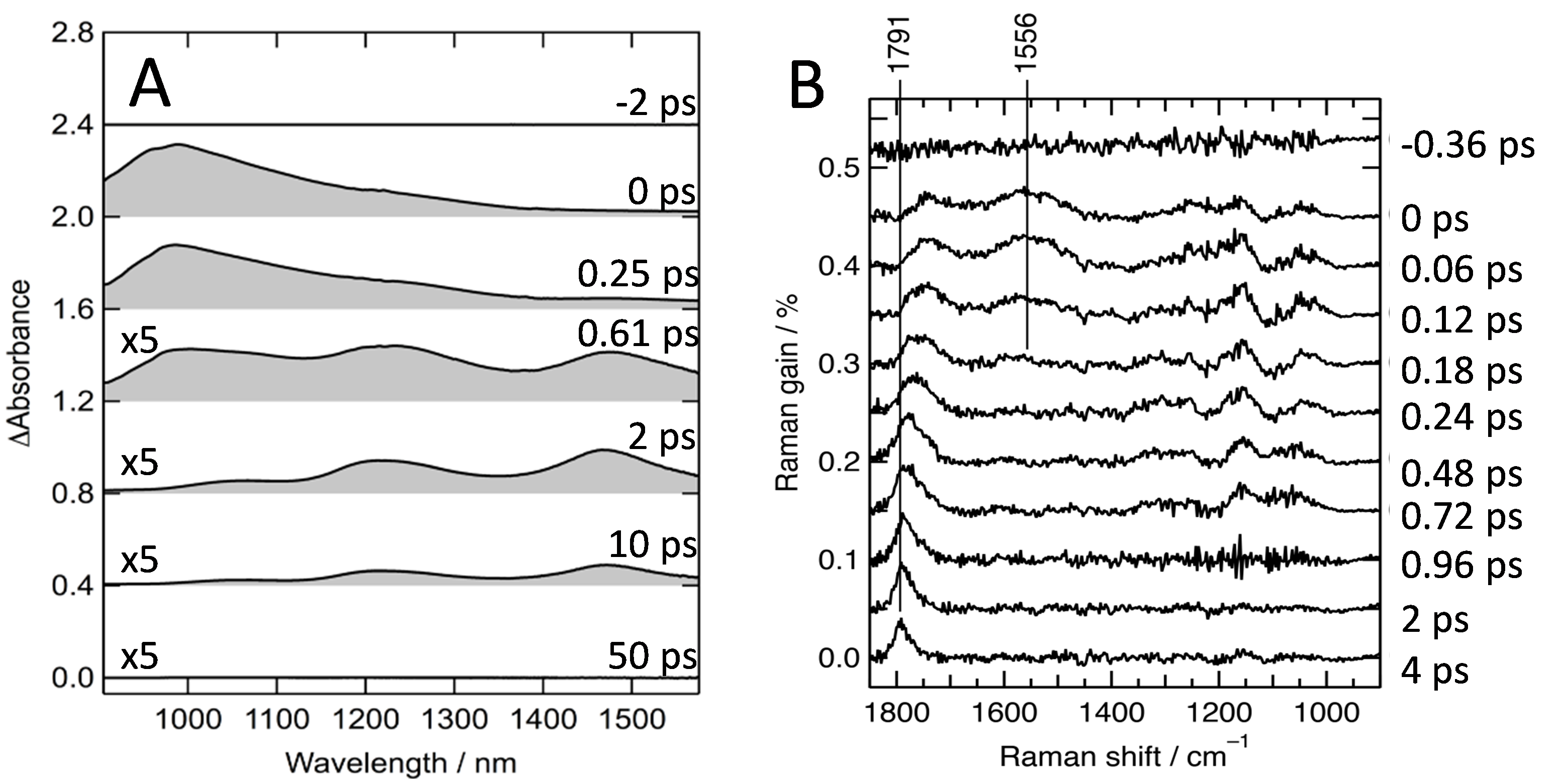
図5 シクロヘキサン溶液中でのβ-カロテンの時間分解近赤外吸収スペクトル(A)
および時間分解近赤外誘導ラマンスペクトル(B)
本稿では、筆者らが作ったピコ秒時間分解ラマン分光計、フェムト秒時間分解近赤外吸収分光計、およびフェムト秒時間分解近赤外非線形ラマン分光計について紹介した。測定技術についての記載が多くなってしまったが、測定法を開発することが筆者らの研究の重要な主題になっていることに免じてご容赦いただきたい。 今回とりあげた3種類の分光計に加えて、ピコ秒時間分解けい光分光計もイオン液体や深共融溶媒、あるいは脂質二重膜の研究で大活躍している[14、15]。本稿ではこの分光計に言及する余裕がなくなってしまったが、別の機会にぜひ紹介したい。
謝辞本稿で紹介した新しい分光計は、共同研究者があってはじめて作ることができた。引用した論文のすべての共著者の方々に改めて感謝申し上げる。
参考文献
- T. Tokita, T. Takaya, K. Iwata, J. Raman Spectrosc. 52, 2051 (2021).
- K. Yoshida, K. Iwata, Y. Nishiyama, Y. Kimura, H. Hamaguchi, J. Chem. Phys. 136, 104504 (2012).
- 岩田耕一,分光研究 54, 153 (2005).
- R.G.W. Norrish, G. Porter, Nature, 164, 658 (1949).
- G.C. Pimentel, Appl. Opt. 7, 2155 (1968).
- K. Iwata, H. Hamaguchi, Appl. Spectrosc. 44, 1431 (1990).
- T. Takaya, H. Hamaguchi, K. Iwata, J. Chem. Phys. 130, 014501 (2009).
- T. Takaya, K. Iwata, J. Phys. Chem. A 118, 4071 (2014)
- M. Ye, K. Iwata, Chem. Lett. 48, 422 (2019).
- M. Kajita, T. Takaya, K. Iwata, Phys. Chem. Chem. Phys. 24, 5418 (2022).
- 「ラマン分光法」,日本分光学会分光法シリーズ1,濵口宏夫,岩田耕一共編,講談社(2015).
- T. Takaya, K. Iwata, Analyst 141, 4283 (2016).
- T. Takaya, M. Anan, K. Iwata, Phys. Chem. Chem. Phys. 20, 3320 (2018).
- 岩田耕一,木村佳文,現代化学474, 23 (2010).
- K. Iwata, M. Terazima, H. Masuhara, BBA General Subject 1862, 335 (2018).